Unravelling the Ovarian Tumour Immune Microenvironment
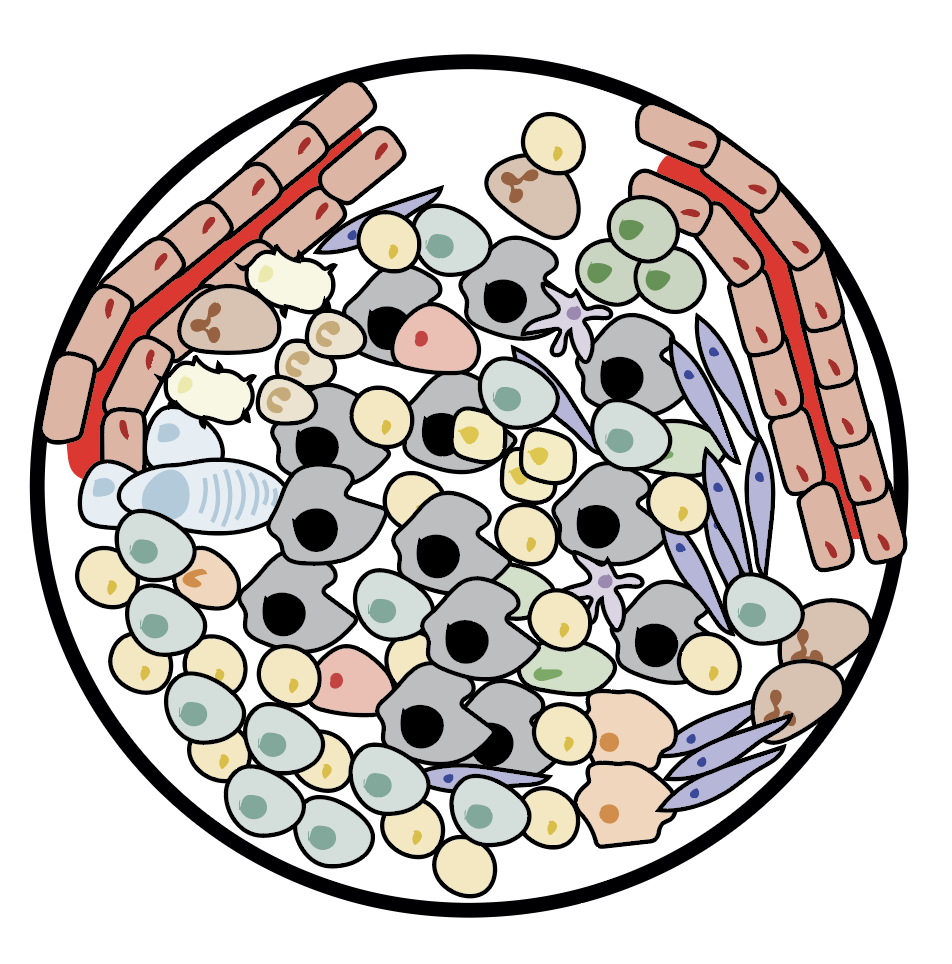
We show ubiquitous tumour-immune microenvironmental variability and we uncover an unanticipated immunogenic effect of chemotherapy (Jimenez-Sanchez et al, 2020, Nat Genet)

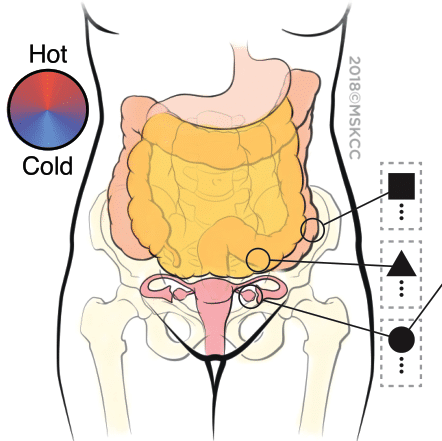
Metastatic Snapshots from an Ovarian Cancer Patient
Distinct tumour immune microenvironments co-exist within a single individual and may help to explain the heterogeneous fates of metastatic lesions often observed post-therapy (Jimenez-Sanchez, 2017, Cell)

Welcome to the Miller lab website
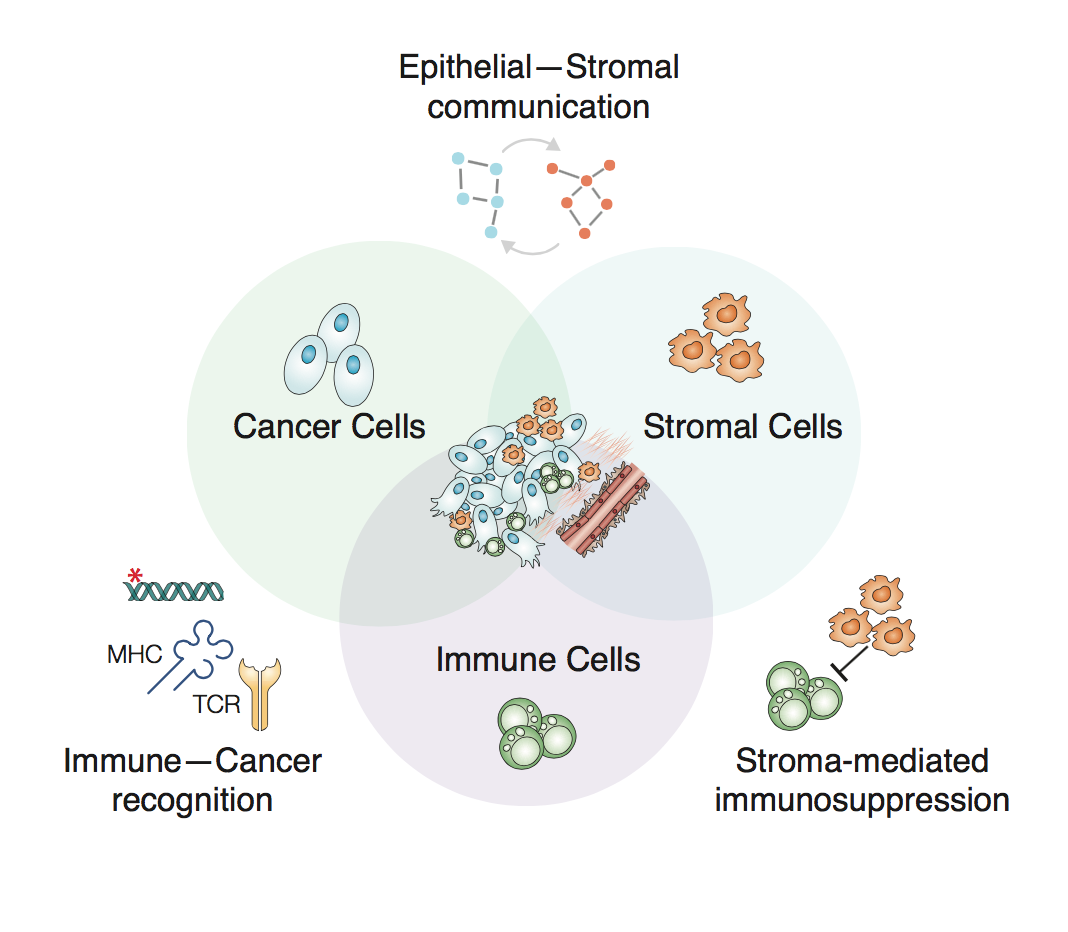
The Cancer Systems Biology group (Miller lab) at CRUK Cambridge Institute uses experimental and computational approaches to characterise cancer signalling and cell-cell interactions